












中国科学技术大学附属第一医院的翁建平教授团队在《Signal Transduction and Targeted Therapy》期刊发表题为“Targeting protein modifications in metabolic diseases:molecular mechanisms and targeted therapies”的综述,深入介绍了多种蛋白翻译后修饰在糖尿病、肥胖、脂肪肝、高脂血症等多种代谢性疾病中参与的生理过程和发生机制,并对这些翻译后修饰的蛋白干预药物在临床中最新的潜在应用与进展展开讨论。
研究背景
代谢性疾病是非传染性疾病最常见的形式,它可通过心血管疾病对全年龄段人的健康造成影响及危害。
蛋白翻译后修饰(PTM)是指在翻译后的蛋白质氨基酸残基上通过添加或移除特定的基团进行化学修饰,从而调节蛋白质的活性、定位、折叠、以及蛋白与其他生物大分子间相互作用。这种调控方式对许多重要的生命活动、疾病发生都有重要影响。迄今,已鉴定出600多种PTM,其中蛋白质磷酸化、甲基化、乙酰化、泛素化、糖基化等是最常见的PTM,此外琥珀酰化、巴豆酰化、2-羟基异丁酰化、乳酸化等是近年来发现的一系列新型酰化修饰,也是近些年来研究的热门方向。
在这篇综述中,作者通过关注基于PTM的蛋白质修饰,对代谢性疾病中涉及的蛋白质和通路进行了全面描述,展示了PTM在临床前研究和临床试验中的药物干预,并提供了未来的观点。

研究内容
磷酸化是将磷酸基团加在中间代谢产物上或加在蛋白质上的过程,是目前研究最深入的PTM之一。磷酸化通常发生在胞质溶胶或细胞核中,被认为是调节蛋白质活性和功能的基本、普遍和基本机制。磷酸化位点可以是丝氨酸、苏氨酸、酪氨酸、半胱氨酸、精氨酸、脯氨酸、天冬氨酸和组氨酸残基,但最常见的靶位点是丝氨酸 (Ser)、苏氨酸 (Thr) 和酪氨酸 (Tyr)。
磷酸化可以通过两种机制快速控制蛋白质的功能,一种是通过变构来激活酶活性(通常是Ser/Thr,Tyr残基),另一种是通过结合相互作用结构域来激活信号转导(通常是 Tyr 残基)。近 30% 的人类基因组编码蛋白含有共价结合的磷酸盐,可逆蛋白磷酸化几乎调节与复制、转录、细胞凋亡、免疫反应、环境反应和细胞代谢等细胞过程的各个方面相关。异常磷酸化已被认为是多种疾病的原因或后果,如肿瘤、心血管疾病和代谢紊乱等。因此,靶向蛋白激酶的药物为针对多种疾病提供了有吸引力的治疗策略。
蛋白质乙酰化是将有机化合物分子中的氮、氧、碳原子上引入乙酰基CH3CO-的反应。乙酰化修饰包括一种不可逆类型(Nα-乙酰化)和两种可逆类型(Nε-乙酰化和O-乙酰化)。这三种类型的乙酰化可以发生在不同频率的不同氨基酸上,赖氨酸 Nε-乙酰化更常见。目前,人类脱乙酰酶有三种类型:HDAC1、HDAC2、HDAC3和HDAC8组成I类HDAC; II 类 HDAC 分为 IIa 类(HDAC4、HDAC5、HDAC7 和 HDAC9)和 IIb 类(HDAC6 和 HDAC10); III 类 HDAC 是含有 SIRT1 至 SIRT7 的烟酰胺腺嘌呤二核苷酸 (NAD + ) 依赖性 sirtuins (SIRT)。乙酰化与乙酰辅酶A水平直接相关。线粒体和非线粒体乙酰辅酶A是独立产生的,可以局部触发乙酰化。最近的研究表明,乙酰辅酶A可以以非酶促的方式调节乙酰化。
细胞核中组蛋白乙酰化和去乙酰化之间的动态平衡改变了染色质结构并调节基因表达。乙酰化刺激染色质解凝聚并促进基因表达,而去乙酰化诱导抑制基因表达。在过去的十年中,日益先进的蛋白质组学信息极大地扩展了已知的乙酰化非组蛋白的数量。已经鉴定出大量非组蛋白乙酰化,并发现与重要的细胞生物学有关,包括基因表达、DNA 损伤修复、细胞周期控制、细胞命运、蛋白质折叠、蛋白质-蛋白质相互作用、自噬、信号转导和细胞代谢。因此,乙酰化的破坏与多种病症和疾病有关,包括免疫紊乱、衰老、肿瘤、神经系统疾病、代谢紊乱和心脏病。
甲基化是将甲基从活性甲基化合物转移到氨基酸残基的催化过程,甲基化主要发生在细胞核中,是一种介导DNA转录可用性的表观遗传调控过程,通常会修饰组蛋白,组蛋白甲基化比其他组蛋白PTM(例如磷酸化和乙酰化)慢得多,表明表观遗传的稳定性。其可修饰几种氨基酸残基,主要修饰赖氨酸和精氨酸残基,赖氨酸残基最多可以甲基化三次,精氨酸残基最多可以甲基化两次,在每次甲基化时,都会从ɛ-氨基中去除一个质子,但这些质子不会影响总电荷,随后会降低氢键能力并增加疏水性。
甲基化与各种细胞活动有关,如转录调控、表观遗传沉默、RNA 加工和输出以及信号转导等。蛋白质甲基化失调会导致多种疾病,如癌症、精神异常、代谢紊乱和心血管疾病等。
泛素化是指泛素分子在一系列特殊的酶作用下,将细胞内的蛋白质分类,从中选出靶蛋白分子,并对靶蛋白进行特异性修饰的过程。
泛素化可以修饰所有 20 种氨基酸,但赖氨酸泛素化是泛素化的主要形式,泛素化是通过泛素-蛋白酶体系统 (UPS) 降解内源性蛋白质的一种公认的机制。UPS 的这一里程碑式发现获得了 2004 年诺贝尔化学奖。一个位点上发生的泛素修饰可以发生单泛素修饰,也可以发生多聚泛素化修饰。多聚泛素化即泛素分子在偶联到特定的蛋白氨基酸后,其它泛素分子可以和现有泛素分子的赖氨酸再进行连接,形成多个泛素的叠加。根据其泛素分子的叠加方式位置,多聚泛素化又分为K11、K6、K48、K29、K63等。目前发现只有特定赖氨酸上的多聚泛素化(主要是K48和K29)与蛋白酶体的降解有关,而其他多泛素化和单泛素化可能调节其他过程,例如内吞运输、炎症、翻译和DNA修复等。
泛素化在干细胞的保存和分化以及各种细胞过程中具有重要意义,如细胞增殖、DNA修复、复制、转录、蛋白质降解、自噬和凋亡、先天免疫和信号转导等。泛素化功能障碍与各种疾病密切相关,如肿瘤、代谢性疾病、炎症性疾病和神经退行性疾病等。
SUMO 蛋白是一种 10kDa 的多肽,通过异肽键与赖氨酸残基的ɛ-氨基连接,这称为SUMOylation。SUMO蛋白在氨基酸序列中与泛素具有相似性(小于20%)。所有SUMO蛋白的N末端共享10-25个氨基酸的无形序列,而泛素化蛋白则不存在该序列。SUMO家族在不同的物种中各不相同,酵母中有两种亚型,植物中有八种亚型,哺乳动物中有三种亚型,人类中有四种亚型。SUMO化与许多生物过程相关,如染色质组织、DNA修复、转录控制、大分子积累、细胞周期进程、运输、基因表达和细胞信号通路等。SUMO化异常在肿瘤、阿尔茨海默病、帕金森病、心血管疾病和代谢紊乱等疾病中被报道。
6.拟泛素化(Neddylation)
拟泛素化是近年来发现的一种类似泛素化的蛋白质翻译后修饰过程,涉及泛素样分子NEDD8与蛋白质赖氨酸残基的共价结合,是蛋白质调控的重要机制。拟素化过程由NEDD8激活酶 (NAE1)、NEDD8连接酶和E3连接酶催化,NEDD8激活酶将NEDD8活化为共价活性形式,NEDD8连接酶负责将NEDD8转移到蛋白质赖氨酸残基上,E3连接酶识别并选择目标蛋白,并促进NEDD8的共价结合。
拟泛素化广泛存在于真核细胞中,参与各种细胞过程,包括细胞分裂、细胞凋亡、信号传导和细胞粘附。异常的类泛素化涉及各种疾病,如肿瘤、神经退行性疾病、代谢疾病和心血管疾病。
7.糖基化(Glycosylation)
糖基化被认为是最丰富和最复杂的PTM,糖基化是在糖基转移酶作用下将糖转移至蛋白质并和蛋白质上的氨基酸残基形成糖苷键,有O-糖基化和N-糖基化。
N-糖基化是最常见的糖基化,其通过β1-糖苷键将N-乙酰氨基葡萄糖(GlcNAc)连接到保守基序Asn-X-Ser/Thr上。N-糖基化包括三个过程:N-糖合成、转移和修饰。N-甘氨酸的生物合成和转移仅发生在内质网中,但修饰可发生在内质网和高尔基复合物中。糖基化早期是个可逆过程,N-聚糖酶 (PNGase) 特异性水解天冬酰胺 (Asn) 连接的寡糖,介导去糖基化。
O-糖基化将 GlcNAc 和 N-乙酰半乳糖胺 (GalNAc) 连接到羟基中氧原子中的丝氨酸或苏氨酸,O-糖基化通常发生在高尔基体的翻译后。O-聚糖由保守的O-GlcNAc转移酶形成,并被高度保守的O-glcNAcase可逆分解,O-连接糖基化在粘蛋白的合成中至关重要。
糖基化在调节细胞过程方面至关重要,包括蛋白质折叠、降解、分泌、分子运输和清除、细胞粘附、细胞间相互作用、信号转导、受体激活和内吞作用。糖基化的失调会影响人类疾病的发展,包括肿瘤、动脉粥样硬化、糖尿病、肝硬化和阿尔茨海默病。
8.瓜氨酸化(Citrullination)
瓜氨酸化又称脱亚胺化,是将精氨酸转化为瓜氨酸的不可逆化学过程,在此过程中,带正电荷的精氨酸被化学水解为不带电荷的瓜氨酸和中性尿素。
瓜氨酸化修饰可涉及许多细胞蛋白,包括细胞核、细胞质、线粒体和细胞膜中的蛋白等。肽酰基精氨酸脱亚胺酶(PAD)的活性和平衡在瓜氨酸化和细胞过程中发挥作用,包括蛋白质稳定性和结构、蛋白质-蛋白质相互作用、细胞凋亡和细胞死亡。瓜氨酸蛋白异常可导致多发性硬化症、癌症、类风湿关节炎、系统性红斑狼疮、阿尔茨海默病和代谢紊乱等。
ADP ribosylation是将ADP核糖(ADPr)从NAD+转移到靶蛋白并释放烟酰胺(Nam)。ADP核糖基化调节主要的细胞过程,包括DNA修复、细胞生长和分化、细胞代谢、应激反应和免疫adp核糖基化失调可导致人类疾病,包括癌症、缺血-再灌注样组织损伤、心脏病、神经系统疾病和代谢紊乱等。
琥珀酰化为一种2010年发现的PTM,是指琥珀酰基团供体(如琥珀酰辅酶A)通过酶学或者非酶学的方式将琥珀酰基团共价结合到底物蛋白质的赖氨酸残基的过程。琥珀酰辅酶a是三羧酸(TCA)循环中重要的代谢中间体,同时也是琥珀酰化的重要供体,使琥珀酰化控制细胞代谢和信号转导,越来越多的证据表明,蛋白质琥珀酰化参与了转录修饰、免疫反应和细胞代谢,包括TCA循环、尿素循环和脂肪酸代谢,代谢改变。目前的资料显示琥珀酰化功能障碍可导致许多疾病,如炎症性疾病、结核病、缺血再灌注样组织损伤和代谢疾病等。
11.巴豆酰化(Crotonylation)
巴豆酰化为一种2011年发现的组蛋白修饰种类,主要发生在组蛋白的赖氨酸残基,是指在巴豆酰辅酶A的作用下,将巴豆酸转化为巴豆酰辅酶A的过程(Kcr)。Kcr通常存在于转录活性染色质区域的组蛋白上,与生殖调控密切相关,近年来的研究表明,其也可发生在非组蛋白上。赖氨酸巴丁酰化与许多细胞过程有关,包括DNA损伤和修复、干细胞分化、精子发生和炎症。赖氨酸巴丁酰化失调与多种人类疾病有关,包括肿瘤、神经精神疾病和心血管疾病等。
12.乳酸化修饰(Lactylation)
乳酸化修饰为一种2019年发现的新型翻译后修饰,是指在乳酸辅酶A的作用下在赖氨酸残基上连接一个乳酸基团,因此也称为赖氨酸乳酸化 (Kla)或乳酸化。乳酸化修饰与多种生理病理过程相关,如细胞分化、M1巨噬细胞极化、线粒体膜结构和功能的稳定性等,同时参与肺纤维化、非酒精性脂肪肝、肾癌、糖尿病、心血管疾病和神经系统失调等疾病的发生进展,等等相关的PTMs。
二、疾病中的PTMs
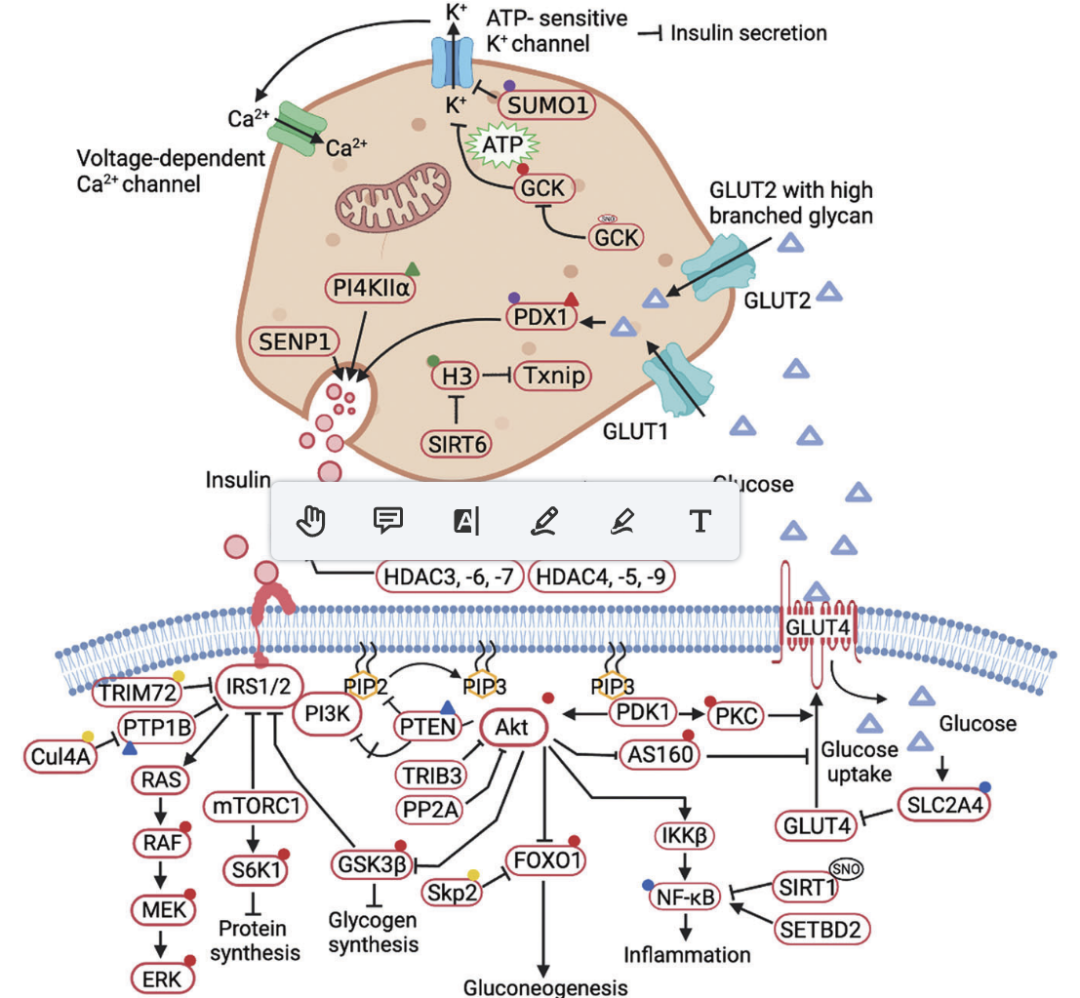
1.1 磷酸化(Phosphorylation)与糖尿病:蛋白磷酸化是一个重要的PTM,它平衡了激酶和磷酸酶在调节葡萄糖刺激胰腺细胞胰岛素分泌中的作用,葡萄糖稳态主要依赖于蛋白激酶和磷酸酶介导的信号级联反应,它们通过控制不同底物的PTMs来决定代谢过程的输出。
1.2 乙酰化(Acetylation)与糖尿病:蛋白质的乙酰化是一种可逆的PTM途径,通过特定类型的酶的功能来调节,这一过程是人体代谢的主要调节剂。乙酰化是一种依赖于乙酰化酶和去乙酰化酶分别催化乙酰化和去乙酰化过程的PTM。乙酰辅酶a为乙酰化提供乙酰基,是糖异生、TCA循环和糖酵解的重要组成部分。因此,在乙酰化与高血糖和代谢系统的胰岛素抵抗之间建立了联系。
1.3 甲基化(Methylation)与糖尿病:组蛋白甲基化和非组蛋白甲基化都是被称为甲基化的PTM类型,蛋白质甲基化主要发生在赖氨酸或精氨酸残基上,并与1至3个甲基连接胞浆中的N -甲基转移酶。蛋白质甲基化通常与基因抑制或激活有关,这取决于修饰的程度和位置。
1.4 泛素化(Ubiquitination)与糖尿病:抑制泛素激活酶E1阻断胰岛素信号关键分子的泛素化,防止棕榈酸诱导的胰岛素抵抗2型糖尿病患者肌肉组织中UBC9蛋白表达降低单倍体充足的UBC13可以改善hfd诱导的胰岛素滞留泛素偶联酶E2O (UBE2O)在肥胖T2DM患者中显著上调。
1.5 类泛素化(Sumoylation)与糖尿病:伴有严重胰岛素抵抗的2型糖尿病患者骨骼肌中UBC9蛋白表达较低时,胰腺β细胞中独特的SUMO偶联E2酶UBC9缺失的小鼠会自发发展为糖尿病,β细胞死亡是活性氧积累的结果,glii -similar 3 (Glis3)是胰岛素调节相关转录因子,pias家族蛋白和UBC9可以介导Glis3下调胰岛素转录。SUMO解偶酶SENP1参与T2DM患者的胰岛素分泌和T1DM患者的脂肪细胞炎症。SENP1定位于β细胞中的胰岛素颗粒,而在β细胞中缺失SENP1小鼠的β细胞糖耐量受损脂肪细胞特异性SENP1的缺失加重了NF-κB必需分子(NEMO)的sumo化和T1DM的症状。
1.6 拟素化(Neddylation)与糖尿病:Cullin蛋白拟素化修饰是由NEDD8- E1、E2和E3酶介导的一个过程,该过程将NEDD8依次转移到Cullin蛋白上,抑制Cullin类化修饰可迅速减少小鼠肝脏葡萄糖生成,减轻高血糖并改善肝脏胰岛素信号,Cullin 3的 RING E3泛素连接酶功能障碍引起糖尿病患者血管收缩和钠重吸收增加。
1.7 糖基化(Glycosylation)与糖尿病:小鼠Glut-2的N -糖基化降低和GlcNAcT-IV表达与高脂肪饮食诱导的糖尿病有关。GlycA被认为是源于N -乙酰氨基葡萄糖的全身性炎症的标志物,全身性炎症可能通过引起胰岛素抵抗和β细胞功能障碍而促进T2DM的发生,补充唾液酸或唾液酸前体N -乙酰- D -甘露糖胺可以恢复抗炎特性并保持胰岛素敏感性。在其他类型的糖尿病中,聚集的N -聚糖是HNF1A-MODY的一种新生物标志物,人乳铁蛋白和分泌性免疫球蛋白A的N -聚糖在妊娠糖尿病中发生了改变等。
1.8 棕榈酰化(Palmitoylation)与糖尿病:蛋白质棕榈酰化被定义为棕榈酸分子通过硫酯键可逆地附着在半胱氨酸残基上的过程。棕榈酰化与胰岛β细胞的代谢失调有关。先前,在人类β细胞系中,GTPase ARF小家族成员ARL15基因的缺失减少了胰岛素分泌。ARL15位于高尔基网络中,表明棕榈酰化依赖高尔基的作用,磷脂酰肌醇4-激酶II- α (PI4KIIα)棕榈酰化积极促进胰岛素信号传导。
1.9 豆蔻酰化(Myristoylation)与糖尿病:肉豆蔻酰基化是由NMTs催化的一种重要的脂肪酸酰化,它可以在蛋白质的氨基末端甘氨酸残基上添加肉豆蔻酰基。STZ诱导的糖尿病动物肝脏NMT活性增加两倍,正钒酸钠可使STZ诱导的糖尿病大鼠肝脏NMT活性恢复正常,此外,肝脏NMT活性与血浆胰岛素水平成反比,但糖尿病对NMT的影响尚不清楚。
1.10 瓜氨酸化(Citrullination)与糖尿病:瓜氨酸化是由PAD酶家族催化的。最近的一项研究表明,每天皮下注射BB-Cl-amidine(一种泛pad抑制剂)可以防止NOD小鼠发生糖尿病,此外,炎症引起的瓜氨酸化几乎只发生在胰腺,可以被认为是β细胞功能障碍或T1D的标志。
1.11 ADP-核糖基化(ADP ribosylation)与糖尿病:ADP核糖基化是由酶催化ADP核糖从NAD+转移到靶蛋白上的PTM,这种PTM通常发生在半胱氨酸、精氨酸和天冬酰胺残基上。PARP在体内和体外被证明是糖尿病和糖尿病并发症的致病标志物。高血糖使糖尿病患者PARP活化增加,降低GAPDH和α-烯醇化酶活性,NAD+被用作ADP核糖基化的底物。活化的PARP可引起NAD+的耗竭,从而破坏胰岛细胞。PARP基因的破坏完全保护了小鼠免受糖尿病的侵害。
此外与糖尿病相关的PTMs还有豆蔻酰化(Myristoylation)、异戊烯化(Prenylation)、谷胱甘肽化(S-glutathionylation)、亚硝基化(S-nitrosylation)、二硫键水解(Sulfhydration)、羰基化(Carbonylation)等。
2. 肥胖症
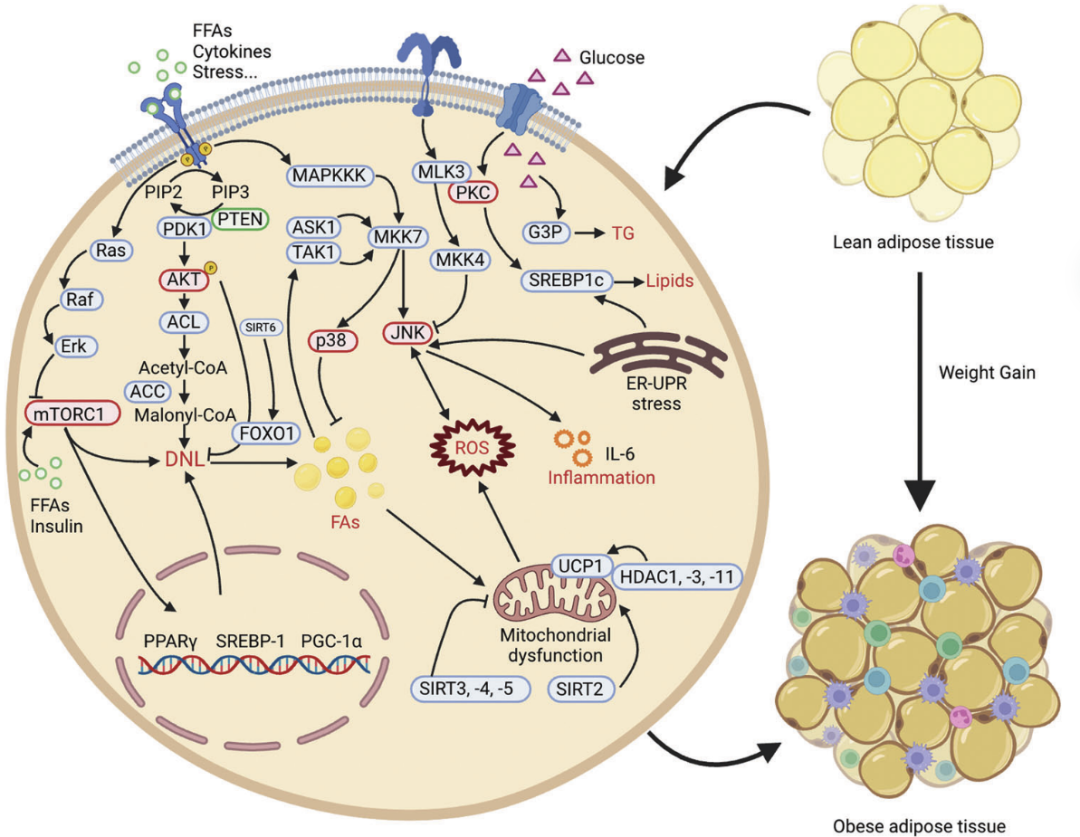
2.1 磷酸化(Phosphorylation)与肥胖症:肥胖是一种代谢性疾病,其最简单的表现是能量摄入和消耗之间的不平衡。慢性营养过剩导致脂肪细胞肥大,从而进一步促进肥胖相关疾病。蛋白激酶通过磷酸化调节许多生物过程。人类gsk3 β-过表达小鼠由于脂肪量增加而体重增加。失活的GSK3β通过Dyrk1A磷酸化抑制脂肪生成蛋白的表达,可能在肥胖的病理过程中发挥作用。MAP激酶相互作用的丝氨酸/苏氨酸激酶1 (MNK1)或MNK2的缺乏可以防止HFD诱导的体重增加等。磷脂酰肌醇-3,4,5-三磷酸[PtdIns(3,4,5)P3]是最重要的磷酸肌醇(PIs)之一,Akt是最知名的靶点。AKT的磷酸化由PtdIns(3,4,5)P3催化。AKT-依赖性FOXO1磷酸化在DIO小鼠中降低。MARK4 (ampk相关家族成员)的缺乏会增强胰岛素刺激的AKT磷酸化,从而激活棕色脂肪,从而减少饮食引起的肥胖。肥胖的潜在原因可能是细胞脂质和葡萄糖失衡或失调等。需要进一步研究蛋白磷酸化,以确定其作为治疗肥胖相关代谢疾病的靶点的作用。
2.2 乙酰化(Acetylation)与肥胖症:蛋白质乙酰化与肥胖尤其相关。蛋白质乙酰化是多种代谢反应的组成部分,如葡萄糖代谢、TCA循环和脂肪酸途径。白色脂肪组织(WAT)、棕色脂肪组织(BAT)和米色脂肪组织的动态调控在很大程度上影响机体肥胖。肥胖小鼠肝脏中,组蛋白H3赖氨酸9和18乙酰化在Tnfα和Ccl2基因上的水平上调等。蛋白质乙酰化、能量代谢和肥胖密切相关。
2.3 甲基化(Methylation)与肥胖症:一项肥胖研究量化了饮食诱导肥胖小鼠的组蛋白甲基化。研究发现4个谷氨酸甲基化位点和1个组氨酸甲基化位点,差异均有统计学意义。其中H2A E67me1和H4 E74me1可能与肥胖的病理过程有关。在H3K27的甲基化过程中,zeste同源物2的增强子(enhancer of zeste homolog 2, EZH2)通过催化H3K27的三甲基化促进脂肪分化、体重和脂肪组织质量。端粒沉默-1 like(DOT-1L)的破坏通过促进H3K79甲基化,特别是H3K79me2修饰来调节bat选择性基因程序。热源性脂肪细胞中DOT-1L的缺失可以保护小鼠免受饮食诱导的肥胖等。
2.4 泛素化(Ubiquitination)与肥胖症:在肥胖中,脂肪细胞分化是由许多转录级联控制的。PPARγ是一种核受体,通过将脂肪细胞从其前体转化为肥胖和代谢性疾病相关。E3泛素连接酶介导的蛋白泛素化和蛋白酶体依赖的PPARγ降解在肥胖的发展中逐渐显示出明确的机制等。ppar γ介导的硒蛋白S (SelS)赖氨酸150和硒蛋白K (SelK)赖氨酸47-48的泛素化和降解是脂肪细胞分化所必需的。CUL2-APPBP2是cullin-RING成员家族的泛素E3连接酶。CUL2稳定PRDM16蛋白,抑制脂肪细胞产热,并通过催化其多泛素化来对抗饮食诱导的肥胖。有研究表明,肥胖糖尿病患者脂肪组织中脂肪酸结合蛋白4 (FABP4)含量较高。FABP4通过下调PPARγ调节脂肪形成,并减轻小鼠饮食性肥胖的发展。
2.5 类泛素化(Sumoylation)与肥胖症:SUMOylation修饰是可逆的,其中修饰的蛋白质可以被SENPs去SUMOylation,SUMO调控与多种疾病密切相关,sumo特异性蛋白酶SENP1缺乏导致SIRT3的高sumo化,这可以通过增加氧化磷酸化和能量消耗来保护小鼠免受hfmo诱导的肥胖。有研究表明,FGF21缺失小鼠出现脂肪营养不良,体脂减少,这与赖氨酸107的PPARγ SUMOylation有关。Krüppel-like转录因子5 (KLF5)也是脂质代谢的重要调节因子,受SUMOylation控制。
2.6 类泛素化(Neddylation)与肥胖症:在脂肪形成过程中,基于nedd8的PPARγ类化修饰对于偶联和稳定PPARγ至关重要,并提供了一种潜在的针对PPARγ类化修饰的抗肥胖治疗策略。在肥胖动物模型和人类患者中,肥胖中的类黄酮化特征是否具有病理生理相关性还有待研究。
2.7 糖基化(Glycosylation)与肥胖症:N-糖基化影响肥胖相关蛋白的结构和功能,研究表明,中心性肥胖与IgG N -糖基化的改变有关,低热量饮食对IgG N-糖基化有显著影响,O- Glcn酰化是一种营养感知和细胞应激反应,营养摄入过多会导致代谢紊乱,包括肥胖和糖尿病,长期摄入高脂肪饮食会增加大脑动脉和心脏中的O-GlcNAc水平。由此可见,O-GlcNAcylation在DIO和代谢功能障碍中具有一定的作用,O-GlcNAcylation水平由O-GlcNAc转移酶(OGT)和O-GlcNAcase (OGA)决定,OGT以脂肪脂质去饱和为目标驱动肥胖,脂肪细胞OGT的缺失消除了小鼠HFD诱导的贪食和肥胖。
此外,与肥胖症相关的PTMs还有棕榈酰化(Palmitoylation)、豆蔻酰化(Myristoylation)、S-谷胱甘肽化(S-glutathionylation)、巯基亚硝基化修饰(S-nitrosylation)、硫巯基化(Sulfhydration)、ADP-核糖基化修饰(ADP-ribosylation)、羰基化(Carbonylation)、S-亚磺酰化(S-sulfenylation)等。
3. 脂肪肝疾病
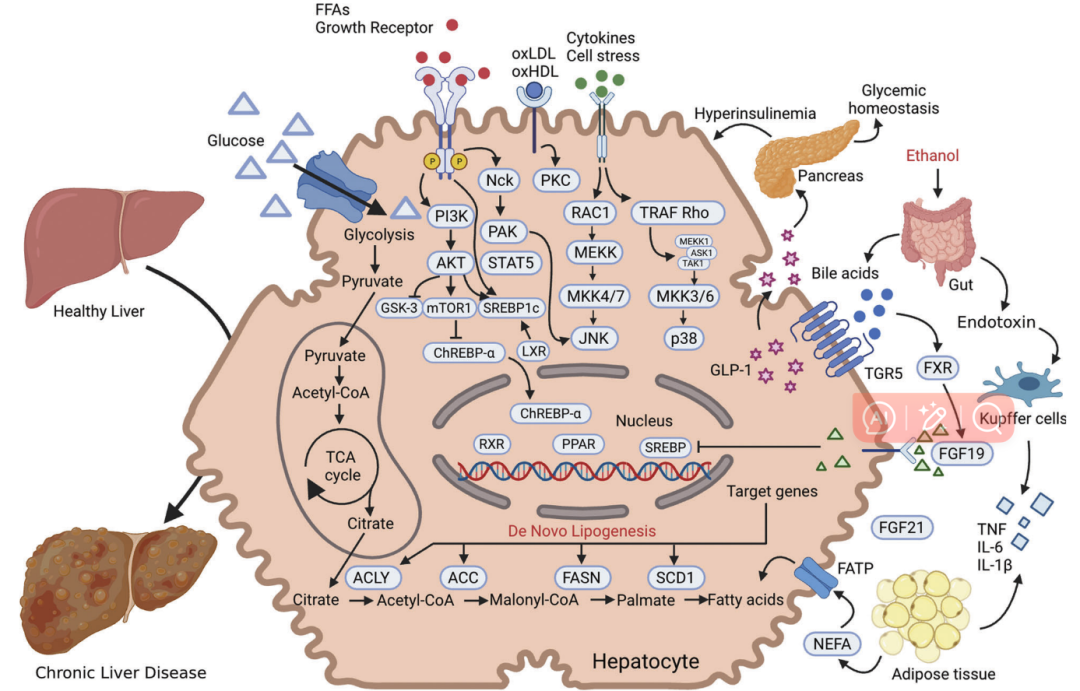
3.1 磷酸化(Phosphorylation)与脂肪肝疾病:蛋白激酶和磷酸酶动态调节蛋白磷酸化。蛋白激酶(PKs)家族包括MAPK、ErbB、PKA-PKD、PI3K/Akt和mTOR,作用于NAFLD的多个下游关键蛋白靶点,调节肝脏糖异生、脂肪生成和炎症。AMPK有多个磷酸化位点,通过磷酸化ACC、SREBPs、GPAT等来调节脂质合成、脂质分解和脂肪酸氧化。ACC可以催化乙酰辅酶a羧化为丙二酰辅酶a,是脂肪酸合成和氧化途径的重要调控位点。ACC的Ser79, Ser1200和Ser1215位点可以被AMPK磷酸化。SREBPs通过磷酸化直接促进参与脂肪酸摄取和甘油三酯(TG)合成的基因。Sn -甘油-3-磷酸酰基转移酶(GPAT)是催化TG合成的速率决定酶。HSL催化TG水解的限速步骤。AMPK通过磷酸化HSL的Ser660和Ser563位点抑制HSL,从而抑制脂肪细胞的脂肪分解等。
3.2 乙酰化(Acetylation)与脂肪肝疾病:蛋白质组学分析已经确定了大量参与中间代谢的乙酰化蛋白。可逆乙酰化是由乙酰转移酶(KATs)和去乙酰化酶(hdac和sirt)控制的。蛋白质乙酰化可调节慢性肝脏的疾病代谢,在NAFL和NASH人类样本中检测到高乙酰化LDHB。锌指蛋白Snail1招募hdac /2诱导H3K9和H3K27去乙酰化,从而抑制脂肪生成。HDAC3控制肝脏脂肪生成的昼夜节律等。
3.3 甲基化(Methylation)与脂肪肝疾病:组蛋白去甲基化酶Jumonji结构域蛋白2B (JMJD2B)去除LXR反应元件(LXREs)附近的组蛋白标记(H3K9me2和H3K9me3),在肝脏X受体α (LXRα)介导的脂肪生成中发挥作用,并参与肝脏脂肪变性。此外,组蛋白去甲基化酶植物同源结构域指2 (Phf2)调节碳水化合物反应元件结合蛋白(ChREBP)上的H3K9me2去甲基化,以防止NAFLD的进展等。
3.4 泛素化(Ubiquitination)与脂肪肝疾病:泛素化是应对慢性肝病中异常折叠或受损蛋白的重要PTM,发挥着多种功能。E3 Ub连接酶和dub在蛋白质泛素化和去泛素化过程中发挥重要作用。激活E3泛素连接酶Ubr1,诱导Plin2多泛素化,防止小鼠肝脏脂肪变性。E3泛素连接酶-含三方基元蛋白31 (TRIM31)通过k48连锁多泛素化促进Rhbdf2降解,缓解小鼠肝细胞NAFLD。敲除Kindlin-2可通过Skp2 E3连接酶依赖性泛素化促进Foxo1的泛素化和降解,从而破坏Foxo1的稳定性,从而保护脂肪肝。Ring finger protein 5 (RNF5)可直接与HRD1结合,促进泛素化降解,抑制NASH进展。此外,分类连接蛋白8 (SNX8)促进FASN蛋白蛋白酶体降解,并通过募集含有28的E3连接酶tripartite motif (TRIM28)来保护NAFLD。E3 Ub连接酶TRIM8和TRIM16可直接结合TAK1,促进其磷酸化,激活JNK/p53和NF-κB信号。TRIM8和TRIM16可减轻NASH患者肝脏脂肪变性和纤维化。E3连接酶FBXW5介导ASK1泛素化并加重NASH等。
3.5 类泛素化(SUMOylation)与脂肪肝疾病:越来越多的证据表明,SUMOylation与肝脏疾病的进展密切相关。例如,UBC9是已知唯一参与SUMOylation调节肝纤维化的e2偶联酶。SUMO-1偶联酶UBC9可降低SREBP-1a中两个SUMO化位点的转录活性,从而抑制脂质产生。此外,小泛素相关修饰物(SUMO) E3连接酶在Lys98位点上抑制SREBP1c,增强SREBP1c与PIASγ之间的相互作用,从而调节营养剥夺时肝脏脂质代谢等。
3.6 拟泛素化(Neddylation)与脂肪肝疾病:最近多种机制的研究已在进行,以阐明在脂质代谢中的关键作用。通过赖氨酸11类泛素化降解SRSF3可部分保护小鼠NAFLD,而SRSF3的缺失易导致小鼠肝细胞癌。此外,cullin 3的类化修饰导致NRF2的失调与AGER1的下调和NASH的加重有关。研究表明,在NAFLD临床前模型中,体内拟泛素化可抑制mTOR的激活和诱导含有mTOR相互作用蛋白(DEPTOR)的蛋白dep结构域,从而介导抗脂肪变性作用并促进肝脏脂肪酸氧化等。
3.7 糖基化(Glycosylation)与脂肪肝疾病:最近的研究表明,许多参与糖基化的蛋白在NAFLD的发病机制中起作用。近年来,糖基转移酶8结构域2 (Glt8D2)表达在严重NAFLD患者中升高,O-GlcNAcylation通过抑制胰岛素信号和激活脂肪生成途径参与肝脏代谢。蛋白O-GlcNAcylation在NAFLD和NASH小鼠脂质积累过程中升高。此外,O-GlcNAcylation还可以修饰肝脏中的ChREBP和FXRChREBP是糖酵解和脂肪生成的关键调节因子。O-GlcNAcylation可使ChREBP蛋白水平升高等。
此外,作者还对与脂肪肝疾病相关的PTMs:棕榈酰化(Palmitoylation)、异戊烯化(Prenylation)、谷胱甘肽化(Glutathionylation)、巯基亚硝基化修饰(S-nitrosylation)、硫巯基化(Sulfhydration)、ADP核糖基化(ADP ribosylation)、羰基化(Carbonylation)等做了描述。
4. 高糖血脂症
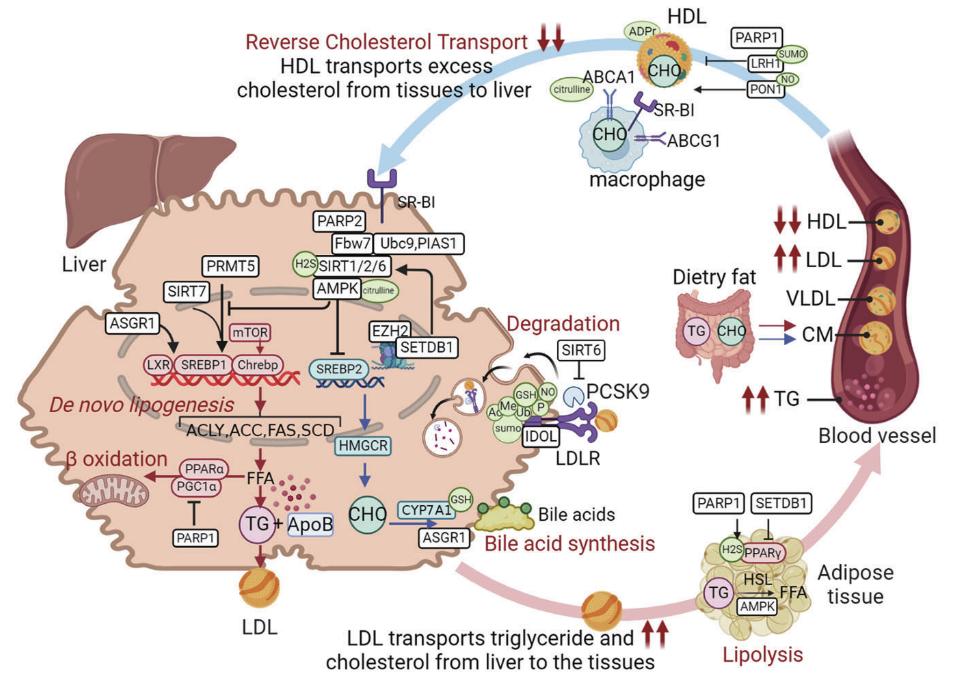
4.1 磷酸化(Phosphorylation)与高糖血脂症:蛋白质磷酸化通过调节脂质分解、脂质生物合成和脂肪酸β氧化中的关键底物来促进脂质代谢,AMPK是一种能量调节激酶,调节几个关键分子参与脂质代谢,如ACC、SREBP等关键酶。ACC是脂肪酸生物合成和氧化途径中具有ACC1和ACC2亚型的重要位点。ACC1在合成途径中介导乙酰辅酶a转化为丙二酰辅酶a的过程,ACC2抑制肉碱棕榈酰基转移酶1 (CPT1)以减少β-氧化。AMPK磷酸化ACC1的第79位丝氨酸,导致ACC1失活和脂肪酸合成减少。AMPK磷酸化ACC2的Ser219位点,导致ACC2的抑制和脂肪酸氧化的增加等。
4.2 乙酰化(Acetylation)与高糖血脂症:乙酰化可以通过改变疏水性、溶解度和表面性质来显著改变蛋白质的功能。Sirtuins (III类hdac)是一组非常保守的NAD依赖的去乙酰化酶,催化与脂质代谢相关的关键蛋白质的去乙酰化反应。Sirtuins被认为通过促进脂肪酸β氧化或输出多余的脂质来抑制脂肪生成和防止脂质积累。SIRT1可使SREBP1c脱乙酰,引起SREBP1c泛素化和降解,抑制脂肪酸和胆固醇的合成。SIRT1诱导PPARα活化并增强脂肪酸β -氧化。SIRT2被认为可以使肝脏中的ACLY去乙酰化381,并抑制脂肪组织中的PPARγ共激活因子1α (Pgc1α) 382,从而抑制脂质积累。SIRT6受SIRT1调控,促进组蛋白H3赖氨酸9乙酰化,抑制TG合成等。
4.3 甲基化(Methylation)高糖血脂症:蛋白质甲基化是许多人类疾病的诱因。甲基转移酶(赖氨酸甲基转移酶、精氨酸甲基转移酶)和去甲基转移酶是调节蛋白质甲基化的酶。赖氨酸甲基转移酶参与脂质代谢。LncRNA PU.1 AS通过与EZH2(一种组蛋白赖氨酸n -甲基转移酶)相互作用,并通过EZH2/SIRT6/SREBP1c途径减少脂肪生成,从而降低血浆TG和肝脏TG等。
4.4 泛素化(Ubiquitination)与高糖血脂症:如转录,翻译和酶活性等多种因素参与脂质稳态。最近有证据表明泛素连接酶在脂质代谢中的作用。在这里,我们将描述E3泛素连接酶的作用。IDOL,个别E3泛素连接酶,通过LXRIDOL -LDLR轴调节LDLR降解诱导高胆固醇血症。IDOL独立于LDLR功能调节循环脂质代谢和动脉粥样硬化的发展。一些E3连接酶调节SREBP。ITCH可通过泛素化降解SIRT6,促进脂肪酸β氧化,ITCH缺乏会干扰核SREBP清除并降低循环胆固醇水平等。
4.5 类泛素化(SUMOylation)与高糖血脂症:SUMOylation是一种由sumo介导的蛋白质修饰,通过影响蛋白质的稳定性、活性、相互作用和细胞定位来影响许多细胞过程;SUMOylation已被确定参与脂质稳态。SUMO调节胆固醇稳态,包括胆固醇合成、摄入、运输和胆汁酸代谢。SUMOylated LRH-1募集协同抑制因子prospero-related homobox protein 1 (PROX1)并抑制与RCT相关的LRH-1依赖基因。FXR是一种胆汁酸刺激的核受体,通过小异源二聚体伴侣(SHP)和LRH-1抑制胆汁酸的生物合成和转运。SUMO1降低了FXR与SHP启动子的附着,抑制了胆汁酸的合成和运输。E2 sumo偶联酶UBC9可以在赖氨酸463位点与SREBP2的SUMOylate相互作用,抑制SREBP2的转录活性并抑制胆固醇合成。活化STAT 1 (PIAS1)蛋白抑制剂,具有SUMO E3连接酶活性,抑制lxr依赖性脂肪酸合成和相关基因等。
4.6 糖基化(Glycosylation)与高糖血脂症:人类脂蛋白的糖基化具有高度的多样性,在调节脂蛋白代谢中起着至关重要的作用。N-糖基化调节参与脂质合成、包装和脂蛋白消除的蛋白质。人体n -糖基化通过增强LDLR表达来降低LDL葡萄糖诱导的N-糖基化维持SREBP裂解激活蛋白(SCAP)的稳定性,减少其与INSIG1的相互作用,允许SREBP激活和下游基因转录。SCARB1编码清道夫受体B类成员1 (SR-BI),是肝脏选择性摄取HDL的主要受体。由于SR-BI降低,SCARB1(亮氨酸取代脯氨酸376)的功能丧失遗传变异诱导n -糖基化改变,增加高密度胆固醇酯并阻止其进入肝细胞等。
4.7 谷胱甘肽化(Glutathionylation)与高糖血脂症:S -谷胱甘肽化是一种重要的氧化还原调节机制,涉及氧化谷胱甘肽和蛋白硫醇通过混合二硫键的附着。在氧化还原调节中,谷胱甘肽经历s -谷胱甘肽化,然后通过酶或化学还原被逆转。谷胱甘肽s -转移酶和过氧化物还毒素促进蛋白s -谷胱甘肽酰化,而谷胱甘肽(Glrx)则主要逆转这一过程。s -谷胱甘肽化及其相关酶在血脂异常中起重要作用。S-谷胱甘肽化可以改变脂质代谢相关的酶和因子。对氧磷酶1 (PON1)是一种与血清HDL相关的酯酶,介导巨噬细胞胆固醇外排,氧化应激可诱导PON1的S-谷胱甘肽化和随后的可逆失活。高脂血症患者谷胱甘肽基血红蛋白水平升高,提示蛋白谷胱甘肽化和氧化应激的作用等。
4.8 巯基亚硝基化(S-nitrosylation)与高糖血脂症:亚硝基化是一氧化氮的亚硝基部分与靶分子之间的共价结合。亚硝基化发生在半胱氨酸的巯基上,称为s -亚硝基化。高脂血症中蛋白质s -亚硝基化的证据很少,主要与脂蛋白调节有关。S-亚硝基化导致半胱氨酸284上的NO和HLD-related PON1相互作用,消除PON1酶活性,导致脂质过氧化物失活和胆固醇外排紊乱等。
此外,作者还对与高糖血脂症相关的PTMs:硫巯基化(Sulfhydration)、瓜氨酸化(Citrullination)、ADP核糖基化(ADP ribosylation)、羰基化(Carbonylation)等做了描述。
5. 动脉粥样硬化
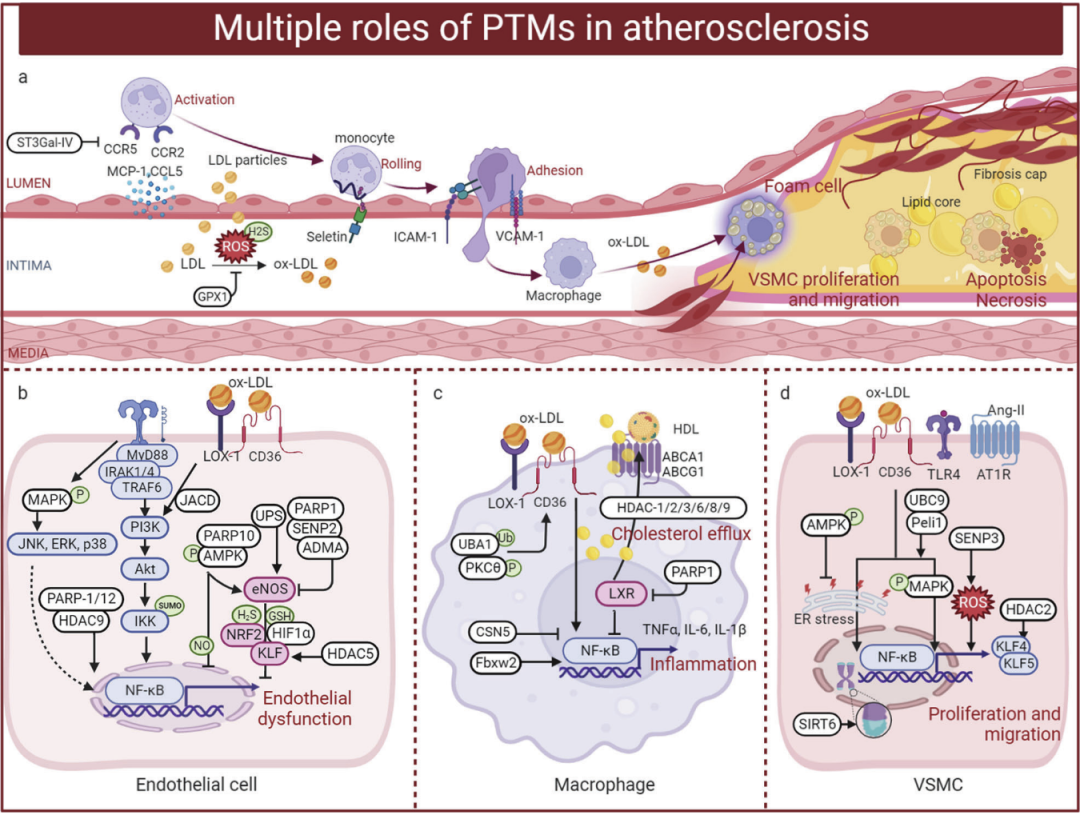
5.1 磷酸化(Phosphorylation)与动脉粥样硬化:蛋白磷酸化是研究最广泛的PTM,它与动脉粥样硬化密切相关。据报道,丰富的蛋白激酶调节动脉粥样硬化的进展,MAPKs是一个丝氨酸/苏氨酸激酶家族,是参与细胞调控的重要信号转导酶,MAPK家族信号级联主要包括p38α MAPK、ERK和JNK.423 MAPK通过基质生成、内皮细胞活化、巨噬细胞炎症和泡沫细胞形成、VSMC增殖和迁移与动脉粥样硬化相关,MAPK活化介导单核细胞粘附活化内皮由氧化LDL((ox-LDL))介导,巨噬细胞p38α MAPK缺失的载脂蛋白E (ApoE-/-)的小鼠显示动脉粥样硬化斑块中巨噬细胞凋亡增强,JNK2降低了清除率受体- a (Sr-A)细胞质尾丝氨酸的磷酸化,使泡沫细胞减少,最终导致ApoE-/- jnk -/-小鼠斑块变小等。
5.2 乙酰化(Acetylation)动脉粥样硬化:JNK2降低了清除率受体- a (Sr-A)细胞质尾丝氨酸的磷酸化,使泡沫细胞减少,最终导致ApoE-/- jnk -/-小鼠斑块变小。赖氨酸乙酰化主要通过调节组蛋白和核蛋白的乙酰化和去乙酰化来影响动脉粥样硬化的过程。ApoE-/-小鼠和高胆固醇血症患者血浆白细胞介素35 (IL-35)升高,IL-35抑制线粒体ro诱导的H3K14乙酰化,抑制内皮细胞活化,缓解动脉粥样硬化的发展等。
5.3 甲基化(Methylation)动脉粥样硬化:蛋白质甲基化已被公认为调节核和核酸结合蛋白的功能,并在心血管疾病中发挥重要作用。组蛋白甲基化在动脉粥样硬化中非常重要。人类动脉粥样硬化病变中的组蛋白甲基化谱显示,在动脉粥样硬化斑块中,H3K27me2和H3K9me2均减少,而在动脉粥样硬化和正常颈动脉中,H3K4me2的水平相当等。
5.4 泛素化(Ubiquitination)动脉粥样硬化:泛素-蛋白酶体级联通路对真核细胞的蛋白质代谢和内源性蛋白质降解至关重要。UPS涉及广泛的生理过程,包括内皮功能障碍、细胞凋亡、氧化应激和泡沫细胞形成,这些都与动脉粥样硬化有关。E1, E2和E3连接酶与动脉粥样硬化进展有关。UBA1是UPS级联中主要的e1激活酶。UBA1抑制剂PYR-41可抑制ox- ldl诱导的巨噬细胞促炎细胞因子表达、NADPH氧化酶和脂质沉积,从而抑制巨噬细胞促炎反应钝化的ApoE-/-小鼠动脉粥样硬化等。
此外,作者还对与动脉粥样硬化相关的PTMs:类泛素化(SUMOylation)、拟泛素化(Neddylation)、糖基化(Glycosylation)、棕榈酰化(Palmitoylation)、豆蔻酰化(Myristoylation)、异戊烯化(Prenylation)、谷胱甘肽化(Glutathionylation)、亚硝基化(S-nitrosylation)、硫巯基化(Sulfhydration)、瓜氨酸化(Citrullination)、ADP核糖基化(ADP ribosylation)、羰基化(Carbonylation)等做了描述。
三、PTMs在临床前研究中的药物干预
1. AMPK活化剂:二甲双胍是一种AMPK激活剂,可以改善葡萄糖控制和胰岛素敏感性,从而减少肠道葡萄糖吸收,二甲双胍促进原代大鼠肝细胞AMPK的磷酸化和葡萄糖的产生。Ampkinone在AMPK的磷酸化中起间接作用,经Ampkinone处理的DIO小鼠体重减轻,脂肪质量下降,代谢特征改善等。
2. MAP激酶抑制剂:SD-169是一种选择性atp竞争性MAP激酶抑制剂。SD-169治疗对NOD小鼠具有降糖作用,并保持β细胞质量,U0126抑制MAPK/ERK通路改善stz诱导的糖尿病小鼠糖尿病性心肌病,U0126还通过抑制肝脏FASN表达来减少脂肪酸的新生合成。U0126抑制ldlr缺陷小鼠动脉粥样硬化,无脂肪生成副作用。
3. PTP1B抑制剂:KY-226是PTP1B的抑制剂,口服KY-226可增加胰岛素诱导的胰岛素受体磷酸化,降低血浆葡萄糖和甘油三酯水平,在高脂肪饮食诱导的肥胖小鼠中,KY-226减少体重增加并增加下丘脑中磷酸化的STAT3;JTT-551已用于T2DM的研究。在db/db小鼠中,长期给药JTT-551显示出抗高血糖作用。单剂量的JTT-551在ob/ob小鼠中增强了肝脏的IR磷酸化;MSI-1436选择性抑制PTP1B并增强胰岛素刺激的胰岛素受体酪氨酸磷酸化,导致DIO小鼠的脂肪特异性体重减轻DPM-1001是MSI-1436的类似物,可以增强β亚基磷酸化,减少饮食引起的肥胖等。
四、靶向蛋白修饰的代谢性疾病的临床试验
越来越多的证据证实了PTMs在代谢性疾病中的重要作用,靶向蛋白修饰酶的药物家族包括激酶激动剂和抑制剂、HDAC抑制剂、组蛋白甲基转移酶抑制剂等。在该篇文章中,作者重点描述了代谢性疾病的药物靶向蛋白修饰的临床试验。
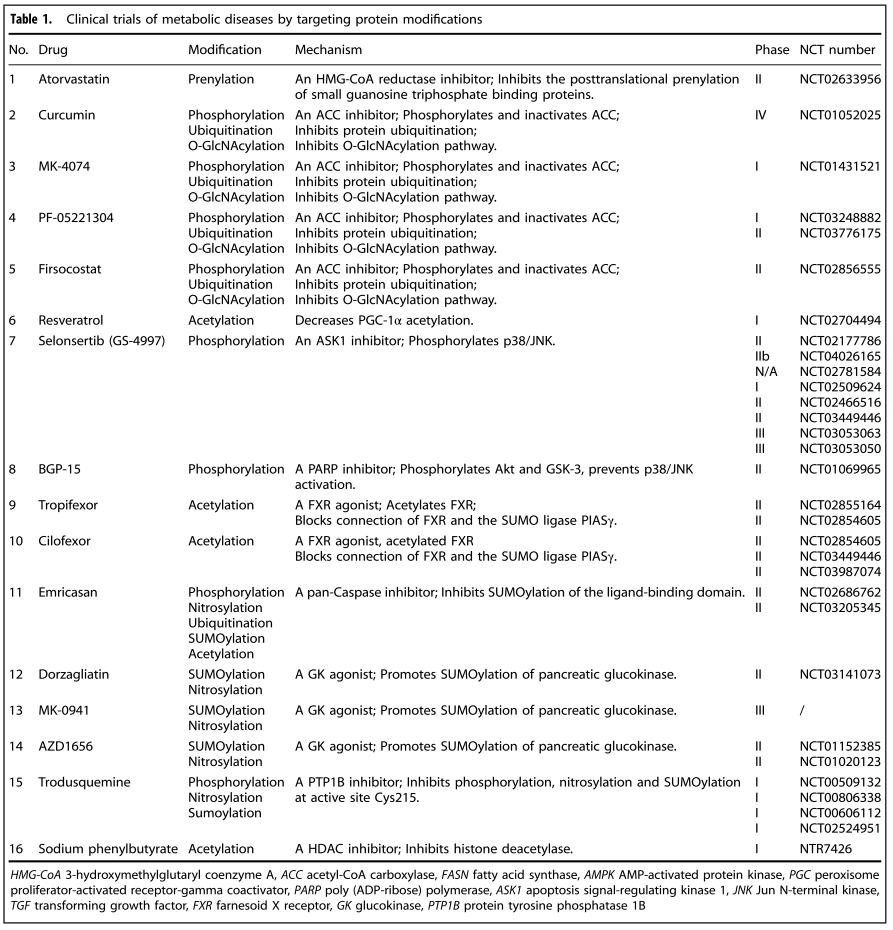
此外文章还描述了PARP抑制剂、FXR受体激动剂、半胱天冬酶抑制剂、葡糖激酶活化剂、PTP1B抑制剂、HDAC抑制剂的临床实验。
五、总结与展望
PTMs控制着靶蛋白的功能和稳定性、蛋白-蛋白相互作用和亚细胞定位,几乎参与了所有的生物过程,从而为系统生物学的调控提供了多种多样的机制,针对PTMS提供了一个新的或精制药物开发的机会,可以为代谢疾病的管理提供一种新的和更精确的方法。

